


Automatic conformer investigator takes tedium out of exploring low-energy chemical space

Quantum chemical calculations drive program that quickly and easily builds up plausible ensembles of molecular conformers
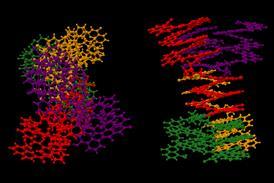
A new program has been developed to suggest the conformations adopted by molecules as they fold and pivot. The program combines a high degree of automation, modest computational demands and broad chemical flexibility, and has already been adopted successfully by experimental and theoretical chemists.